AI-accelerated Docking
Traditional brute-force docking is a powerful tool in early drug discovery, but when applied to ultra-large libraries, it quickly becomes computationally expensive and time-consuming. Our AI-accelerated docking workflow integrates machine learning-driven active learning with classical docking, enabling faster, cost-effective screening of vast chemical spaces. Multiple publications have proven the efficiency of this method in detecting the top-scoring compounds. [1,2,3]
Available Chemical Spaces
- Diverse or targeted subsets from Enamine REAL or Chemspace Freedom Spaces up to 1B
- Custom selection from spaces based on required criteria
- Enamine REAL Database (9.6B molecules)
Why AI-accelerated Docking?
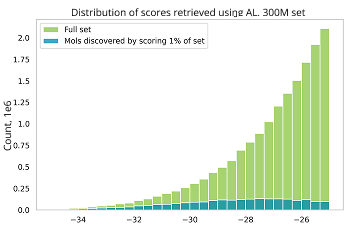
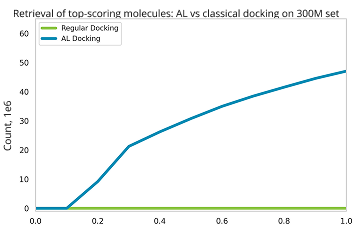
The workflow is designed to handle ultra-large compound collections, where performing classical docking would not be feasible. This allows to explore a larger chemical space to potentially get better hits.
Instead of docking an entire library, we use a model-guided approach to focus only on the most promising regions of chemical space. This reduces computational time by 10–100 times compared to brute-force docking, without compromising the quality of the results.
This workflow has been applied in internal R&D and external collaborations, identifying validated hits for diverse targets under real-world timelines. The efficiency of active learning has been also showcased in multiple publications. [1,2,3]
We provide full project support, including preparation of the chemical space, docking model setup, active learning cycles, and final hit selection. The workflow is fully modular and can be run end-to-end or partially, depending on your needs. You're welcome to be involved in any stage of the process - whether it's reviewing intermediate results or making key decisions - we're happy to work alongside your team.
Project Requirements
For us to complete this workflow on your target of interest, a well-defined protein structure must be available (X-ray, Cryo-EM, NMR, homology models). Additional requirements for compound set pre-filtering can be discussed upon request.
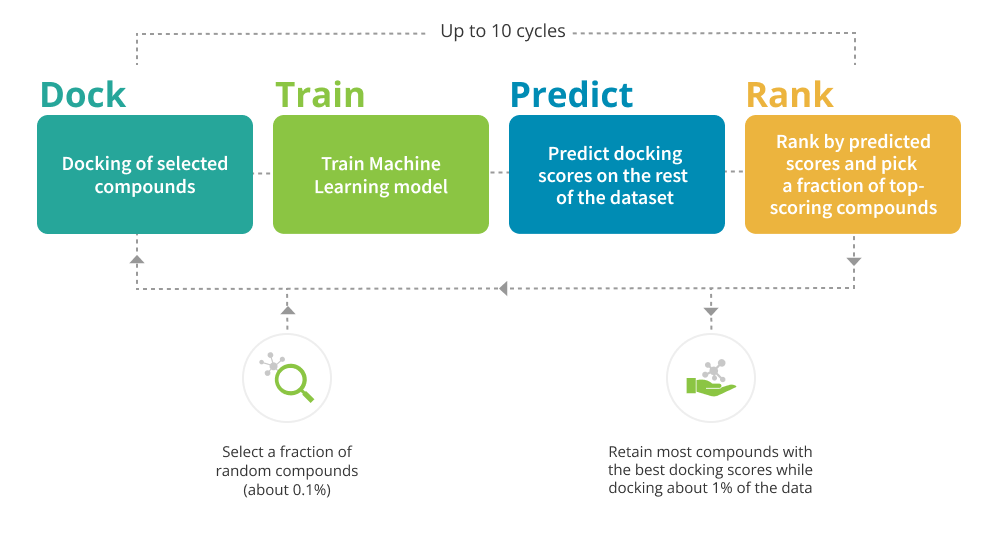
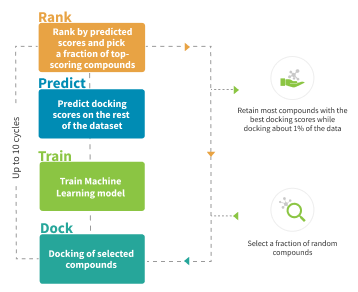
Workflow Description
The project starts with the development of a docking model based on your protein of interest. Our professionals evaluate the quality of the protein structure in order to proceed with docking.
The first fraction of compounds is selected randomly from the chemical space of choice to get a good representation of the chemotypes. These compounds are docked into the target, the resulting scores are used to train a machine-learning model. Using this model, the scores are predicted on the rest of the dataset.
The next compounds for docking are selected based on the predictions of the model – top compounds are picked based on the predicted score.
The process is repeated until most of the top-scoring compounds are retained. Usually, this requires docking of approximately 1% of the library.
As a result, you receive all the docked compounds with the corresponding docking scores.
- Sivula, T.; Yetukuri, L.; Kalliokoski, T.; Käsnänen, H.; Poso, A.; Pöhner, I. Machine Learning-Boosted Docking Enables the Efficient Structure-Based Virtual Screening of Giga-Scale Enumerated Chemical Libraries. J. Chem. Inf. Model. 2023, 63 (18), 5773–5783. https://doi.org/10.1021/acs.jcim.3c01239 .
- Yang, Y.; Yao, K.; Repasky, M. P.; Leswing, K.; Abel, R.; Shoichet, B. K.; Jerome, S. V. Efficient Exploration of Chemical Space with Docking and Deep Learning. J. Chem. Theory Comput. 2021, 17 (11), 7106–7119. https://doi.org/10.1021/acs.jctc.1c00810 .
- Graff, D. E.; Shakhnovich, E. I.; Coley, C. W. Accelerating High-Throughput Virtual Screening through Molecular Pool-Based Active Learning. Chem. Sci. 2021, 12 (22), 7866–7881. https://doi.org/10.1039/d0sc06805e .