Molecular Docking
Molecular docking is an in silico approach utilized in structure-based drug design that allows to predict the binding affinity of a ligand within a binding pocket of a protein target. Scoring functions as well as other output parameters serve as a guide that helps understand the nature of ligand-receptor interaction. Molecular docking in drug discovery presents a resource-effective starting point for early stages of drug development. Chemspace offers several tailored docking approaches suitable for screening subsets of different size.
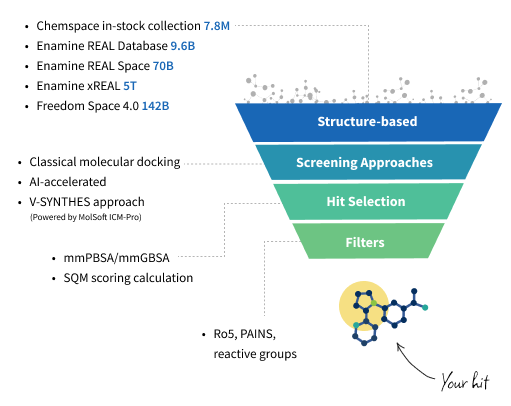
Our Molecular Docking Services
For our in-stock compound collections, we offer classical molecular docking approach. When working with larger subsets (up to 1B), our AL-accelerated docking speeds up the process while maintaining accuracy, making it ideal for mid- to large-scale virtual screens. For ultra-large chemical spaces, including billions of compounds, we utilize V-SYNTHES, an innovative, fragment-based method that enables efficient and accurate screening at scale. The flexibility behind our strategy ensures the most effective solution for your project, regardless of number of molecules in screening subset.
Hit Selection
We also provide a range of methods to refine molecular docking results. For hit selection we utilize post-docking techniques that enhance accuracy and increase the chance to identify true binders.
Semiempirical quantum mechanics (SQM) scoring. We use PM6-D3H4X method, which is often used to estimate protein-ligand binding affinities due to its balance of accuracy and computational efficiency. It can provide reasonable interaction energies (ΔE_int) that correlate well with density functional theory (DFT) results in the gas phase. The change of solvation free energy upon complex formation is calculated using the Conductor-like Screening Model (COSMO2 model) at the PM6 level.
Molecular mechanics Poisson− Boltzmann surface area (mmPBSA) / molecular mechanics generalized Born surface area (mmGBSA) methods. MM/PBSA and MM/GBSA are used to evaluate docking poses, predict binding affinities and hotspots. At the first step the MD simulation of the protein−ligand complex is performed using an explicit solvent mode after which MD snapshots are collected and binding energy is calculated. MM/PB (GB)SA calculations with a proper parameter adjustment (selection of the continuum solvation models and the solute dielectric constant) outperforms classical docking score functions and might considerably improve enrichment rate.
Our Experience
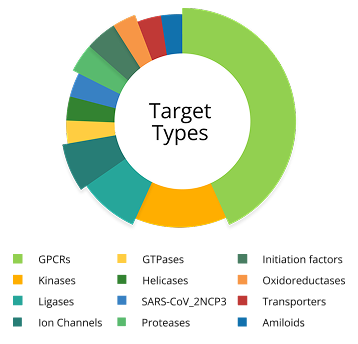
Chemspace’s Molecular Modeling Team has over 5 years of experience applying molecular docking in drug design. During this time we have managed to work with more than 10 different types of targets, including GPCRs, kinases, SARS-CoV-2NSP3, and others. The hit rate typically ranges from 1.3% to 33%, depending heavily on the target type and the availability of existing data.


Why Choose Our Services?
-
Expertise and Experience
Our experienced team in computational biology and bioinformatics uses advanced tools and methods to deliver accurate, reliable results.
-
Integrated Approach
Support fully integrated projects - from hit finding to preclinical studies, utilizing DMTA cycle.
-
Scale
Provide molecular docking for compound sets of any size - from thousands to trillions - tailored to your project needs.
-
Data Security
Ensure your research stays secure and confidential through strict data management practices.
Frequently Asked Questions
Which software tools are utilized in our molecular docking services?
What information is required to initiate a molecular docking project?
How long does a typical molecular docking project take?
How can I request a quote or initiate a project?
What is Molecular Docking?
Molecular docking is one of the approaches that are widely used in structure-based drug design to predict the bond confirmations between small molecule ligands and macromolecule targets. Nowadays it is an important component in the early-stage drug discovery process. Introduced in the 1980s, now molecular docking represents a powerful method due to several key factors:
- Increasing the computer hardware’s power
- Ease of access to the small molecule collections
- Availability of the structural information of the proteins
The core principle of molecular docking is to determine the ‘best fit’ of the ligand (small molecule) into
the binding site of the protein of interest with the help of computer software. It helps researchers to
understand the nature of binding potential drug candidates to the target and dive into the insights of the
drug development.
To perform the screen there are some starting requirements. You need to have a structure for the target.
Traditionally, it can be obtained using such approaches as NMR, cryo-EM, or X-ray crystallography. But on
the other side, we can also get the structure from homology modeling constructions.
Our Methodology
We at Chemspace provide custom docking approaches that are based on the screening subsets of your interest.
- In-stock screening collections of Enamine (4.7M) and Chemspace (7.9M). In that case, we can offer you a classical molecular docking approach for the prediction of protein-ligand interaction.
- AL-accelerated molecular docking of 1 billion subsets created based on your requirements. We utilize machine learning technology to efficiently explore giga-scale chemical spaces.
- V-SYNTHES approach powered by MolSoft ICM-Pro is available for the exploration of Enamine xREAL Space (4.4 trillion molecules). It is a virtual synthon hierarchical enumeration screening approach, that allows screening of the chemical spaces of billions and trillions of compounds without full enumeration.
Thus, Chemspace’s computational docking services are fully customizable and can significantly enhance your drug discovery process.
Importance in Drug Design
Molecular docking is one of the virtual screening approaches. It enables the in sillico evaluation of large-scale chemical collections, efficient identification and optimization of potential drug candidates. The core of this approach is to predict the ligand-receptor binding affinity. It also helps to visualize binding poses and prioritize the compounds for further synthesis and testing. Integration of molecular docking services into drug development process leads to data-driven decisions reducing the time and cost associated with experimental testing.