Virtual Screening
Virtual screening (VS) is a computational approach integral to drug discovery, serving as a
cost-effective and efficient method for identifying potential hits. By simulating molecular interactions
in silico, virtual screening allows researchers to prioritize and filter through numerous compounds,
significantly reducing the number of candidates that need to be experimentally tested [1, 2, 3].
There are two main approaches inside VS: structure-based VS (SBVS) and ligand-based VS (LBVS) [3].
Chemspace provides advanced virtual drug screening solutions, combining structure-based and ligand-based
approaches.
Structure-Based Virtual Screening (SBVS)
SBVS or target-based VS (TBVS) attempts to predict the binding orientation of ligand (organic small
molecule) and target molecule (protein or other biomacromolecule). The main requirement for this
technique is presenting of good enough 3D structure of the target molecule.
For SBVS we offer:
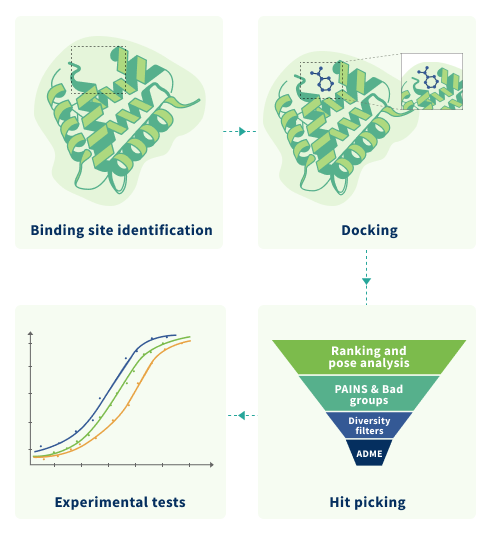
Molecular Docking
Molecular docking is a computational method to predict the preferred orientation and
ligand-target interactions, thus enabling scoring the molecules on how well they fit into the
pocket of the target protein. Top molecules can be identified by the docking score and used in
other computational methods or screened in the lab.
We use ICM-Pro by MolSoft to complete the docking projects. MolSoft software performed well and
outperformed a range of other methods in the numerous blinded modeling competitions and docking
challenges including covalent docking, predicting protein-protein complexes [4].
4D Docking (Ensemble Docking)
Using structure assembles for docking computations we can run 4D docking. This protocol works
twice as fast as regular docking in the number of structures separately and opens the
opportunity to consider the flexibility of the target [4, 5].
RIDGE (Rapid Docking GPU Engine)
RIDGE is a fast and accurate structure-based virtual ligand screening method. Utilizing RIDGE
makes it possible to dock approximately 100 compounds per second or 10M compounds database in
30h. For more details about the engine please find at Molsoft
website
.
3D target-performed pharmacophore modeling
By adding the excluded volumes based on information about ligand surroundings in the ligand-target
complex we improve the classical pharmacophore. This approach outperforms pharmacophore screening,
helps to avoid clashes, and gets more accurate results [6, 7].
To reinforce 3D pharmacophore screening as well as other ligand-based approaches, Chemspace utilizes
cutting edge utilities and one of them is GINGER from Molsoft.
GINGER (Fast GPU Based conformer generation)
GINGER (Graph Internal-coordinate Neural-network conformer Generator with Energy Refinement) is a new
cutting-edge software for lightning-fast high-quality conformer library generation on GPUs [8]. The
productivity of GINGER allows the generation of 10M compounds library in a day and quick processing of
your in-house 2D combinatorial or AI-generated libraries, enabling the application of ligand 3D
structure-based methods such as field-, shape- and pharmacophore-based screening. More details about
GINGER please find
here.
Ligand-Based Virtual Screening (LBVS)
LBVS uses ligands with known biological activity aiming to identify molecules with similar structures.
The main principle is that structurally similar molecules may exhibit comparable biological activity. To
create a ligand-based model for screening the 1D (molecular weight, solubility, or other physicochemical
properties), 2D (molecular topology) or 3D (shape) descriptors could be used. Our virtual drug discovery
approach accelerates early-stage hit identification while reducing costs.
For LBVS we offer:

Pharmacophore modeling
We support 2D and 3D pharmacophore models using the ligand structure information or 3D
coordinates. By utilizing the common chemical features presented only in the most active
compounds we enable the search for similar molecules in the chemical spaces [6].
2D/3D QSAR
Creation of 2D/3D QSAR models based on various methods of linear and nonlinear regressions as
well as using ML tools [4, 9].
Shape-based screening
RIDE is a fast 3D molecular similarity search method based on
Atomic Property Fields (APF) and enabled for Enamine REAL Database (9.6B molecules) and
Chemspace Screening collection (7.8M molecules). Atomic Property Fields (APF) is a grid 3D
pharmacophore potential that is generated from one or more high-affinity scaffolds with seven
properties assigned from empiric physico-chemical components. These properties include hydrogen
bond donors, acceptors, Sp2 hybridization, lipophilicity, size, electropositive/negative, and
charge. APF has also been extended to multiple flexible ligand alignments using an iterative
procedure [4].
Real-time ultrafast shape recognition with pharmacophoric constraints
(USRCAT) – is the new fingerprint for 3D similarity search. We created a workflow
that aims to find compounds with similar molecular 3D shapes to the reference ones, and at the
same time to expand chemical diversity and identify new and potentially active scaffolds [10].
What is Virtual Screening?
Virtual screening is a computational approach focused on identifying promising hit molecules that are
likely to effectively bind to the target protein. Essentially, it is a set of computational algorithms
and models aimed at virtual screening of large chemical libraries to find compounds with high binding
affinity. In modern drug discovery, virtual screening is one of the most commonly used techniques
applied on the early stages. The benefits of virtual screening in drug design are:
- Rapid screening of millions of compounds,
- It can be tailored to the different targets (single proteins or complexes, DNA, RNA, etc.) and
integrated with AI/ML,
- Significant reduction of time and cost by filtering out unpromising chemical compounds.
We specialize in large scale virtual screening, enabling evaluation of millions of molecules from
ultra-large libraries.
Virtual Screening Technologies
Nowadays virtual screening is a cornerstone of the early-stage drug discovery pipeline that allows
scientists to screen ultra-large chemical libraries before costly laboratory testing. Implementation of
state-of-the-art computational approaches into our workflow enables us to reduce costs and at the same
time speed up the research process, staying focused on the active compounds. Virtual screening
approaches include structure-based methods (i.e. our starting point is a finely resolved 3D conformation
of the target) and ligand-based screening (i.e. known active compound is used as a starting point for
further discovery).
Key structure-based methods include:
-
Molecular docking. We perform simulation of the binding process of potential drug candidates to a
target protein to filter out most promising compounds.
-
Molecular dynamics (MD) simulation. It is a computational technique that is aimed at modeling of
flexibility and motion of molecules over time, thus providing insights into dynamics of molecular
systems.
-
Structure-based pharmacophore modeling. Pharmacophore modeling is a frequently used method that
utilizes 3D structure of target to identify a set of essential chemical features, known
aspharmacophore, that are determinants of binding to a selected target protein.
Ligand-based screening approaches include
-
QSAR modeling. This method utilizes statistical and ML techniques to correlate chemical properties
with biological ones.
-
2D/3D similarity search. Different similarity search options help us find new molecules with similar
structural or chemical features to known active compounds.
Benefits of Our Virtual Screening
Chemspace’s virtual screening services provide a cost-effective, fast, and low-risk way to identify
promising drug candidates. By evaluating millions of compounds in silico, there is no need to synthesize
molecules immediately, reducing both time and expense. Our team has access to the ultra-large
combinatorial spaces, including unique in-stock and make-on-demand compounds, thus maximizing diversity
and hit potential. We create a custom workflow to narrow down the most promising candidates for
synthesis with a success rate over 80%.
Frequently Asked Questions
What information is required to initiate a virtual screening project?
As we are aimed to find potential drug candidates for a given target, protein selection of a target
protein and/or the specific domain that is to be inhibitied is the initial step in a virtual
screening project.
How accurate is virtual screening in identifying potential drug candidates?
Virtual screening is a simulation of binding between a ligand and a protein that cannot fully embody
complicated processes that underlie binding in in vitro or in vivo biological system. However, it
certainly helps establishing structure-activity relationship (SAR) as gives an idea of hydrogen
bonding or hydrophobic interactions between a molecule and a protein. Virtual screening hit rates
observed in biological assessment is typically in range of 1% and 20%.
What are the advantages of using virtual screening in drug discovery?
Virtual screening is time- and cost-efficient as at this stage we do not need costly wet laboratory
techniques neither for compound synthesis, nor for binding assays. Hits identified in virtual
screening campaign must be tested in biological set-ups, but the number of output compounds
(100-500) is much lower than for classical HTS campaign (thousands or millions).
How long does a typical virtual screening project take?
The duration of virtual screening project hugely depends on an approach undertaken. Nevertheless,
typical workflow takes approximately 10-12 weeks to accomplish.
What software tools are commonly used in virtual screening?
For molecular docking we use ICM-Pro (Molsoft).
References
-
Slater, O.; Kontoyianni, M. The Compromise of Virtual Screening and Its Impact on Drug Discovery.
Expert Opinion on Drug Discovery 2019, 14 (7), 619-637.
https://doi.org/10.1080/17460441.2019.1604677
-
Kontoyianni, M. Library Size in Virtual Screening: Is It Truly a Number's Game? Expert Opinion on
Drug Discovery 2022, 17 (11), 1177-1179.
https://doi.org/10.1080/17460441.2022.2130244
-
Lavecchia, A.; Giovanni, C. Virtual Screening Strategies in Drug Discovery: A Critical Review. CMC
2013, 20 (23), 2839-2860.
https://doi.org/10.2174/09298673113209990001
-
https://www.molsoft.com/
-
Amaro, R. E.; Baudry, J.; Chodera, J.; Demir, Ö.; McCammon, J. A.; Miao, Y.; Smith, J. C. Ensemble
Docking in Drug Discovery. Biophysical Journal 2018, 114 (10), 2271-2278.
https://doi.org/10.1016/j.bpj.2018.02.038
-
Giordano, D.; Biancaniello, C.; Argenio, M. A.; Facchiano, A. Drug Design by Pharmacophore and
Virtual Screening Approach. Pharmaceuticals 2022, 15 (5), 646.
https://doi.org/10.3390/ph15050646
-
Schaller, D.; Šribar, D.; Noonan, T.; Deng, L.; Nguyen, T. N.; Pach, S.; Machalz, D.; Bermudez, M.;
Wolber, G. Next Generation 3D Pharmacophore Modeling. WIREs Comput Mol Sci 2020, 10 (4), e1468.
https://doi.org/10.1002/wcms.1468
-
Raush, E., Abagyan, R., & Totrov, M. (2024). Efficient Generation of Conformer Ensembles Using
Internal Coordinates and a Generative Directional Graph Convolution Neural Network. Journal of
Chemical Theory and Computation, 20(9), 4054–4063.
https://doi.org/10.1021/acs.jctc.4c00280
-
Kwon, S.; Bae, H.; Jo, J.; Yoon, S. Comprehensive Ensemble in QSAR Prediction for Drug Discovery.
BMC Bioinformatics 2019, 20 (1), 521.
https://doi.org/10.1186/s12859-019-3135-4
-
Kyrylchuk, A.; Kravets, I.; Cherednichenko, A.; Tararina, V.; Kapeliukha, A.; Dudenko, D.;
Protopopov, M. Creation of Targeted Compound Libraries Based on 3D Shape Recognition. Mol Divers
2023, 27 (2), 939-949.
https://doi.org/10.1007/s11030-022-10447-z