Fragment-Based Drug Discovery
Fragment-based drug discovery (or fragment-based drug design) is a powerful method for identifying promising starting points for new drugs. By starting points, we imply small molecules (‘fragments’) that comply with the rule of 3 (Ro3): molecular weight below 300 Da, logP < 3, three or less rotatable bonds, as well as a maximum of three hydrogen bond donors and acceptors each. Starting with a small fragment (12-14 heavy atoms) considerably reduces the size of possible molecules, allowing a fragment library screen containing only 5000 fragments to provide broader coverage of possible chemical and property space to assess the druggability of a new target. While initial hits are typically weak (high micromolar to millimolar in binding affinity), it is considered that fragments can be more suitable for further optimization than hit-like small molecules. At Chemspace we have developed a custom workflow that allows us to identify potential fragments and grow them into hit compounds inside Enamine REAL and Freedom spaces. This approach helps us to avoid the challenging synthesis process of the full compounds.
How Our FBDD Service Works
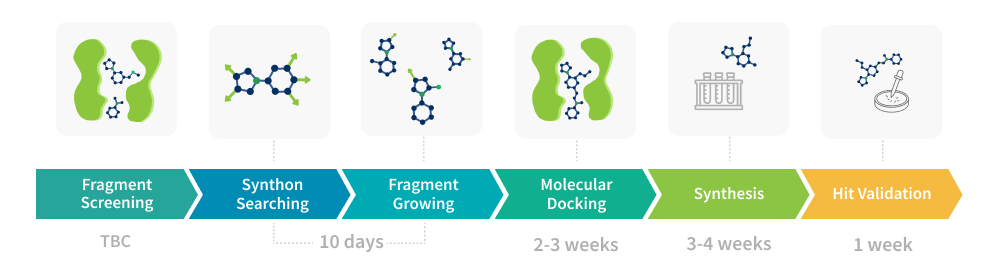
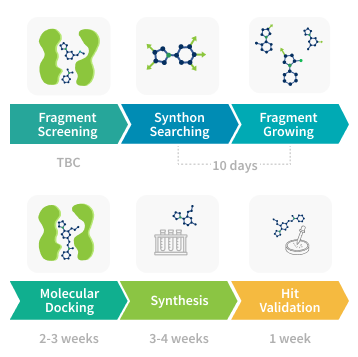
Our fragment-based drug discovery (FBDD) services are tailored to deliver rapid and cost-effective solutions for early-stage hit identification and optimization. Our main goal was fast and efficient fragment growing and merging within Enamine xREAL (4.4T molecules) and Freedom (296B molecules) spaces. It helped us to develop a custom pipeline that efficiently combines FBDD services and commercially available combinatorial spaces. The described approach is cost-effective, since we focus on top-ranked compounds only, thus limiting synthesis to 100-500 most promising molecules.Our platform integrates all the essential stages of fragment drug discovery, including screening, growing, docking, and validation.
-
Fragment Screening
A variety of biophysical methods, such as DSF, SPR, NMR, and X-ray, can be utilized for fragment-based screening. Our team will help you to select the optimal method for your target and enable fragment screening drug discovery using our specifically developed fragment libraries or any other commercially available libraries. The following stages of our FBDD platform can be built up on the confirmed fragment hits resulting from the fragment screening.
-
Synthon Search
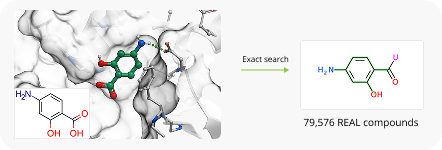
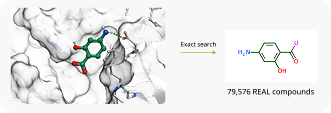
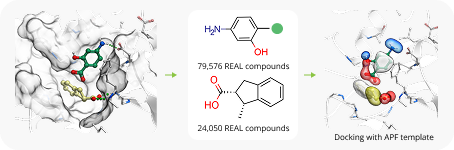
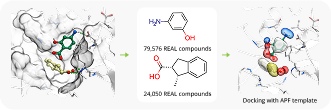
Search synthons with the needed exit vectors in REAL and Freedom spaces.
-
Fragment Growing
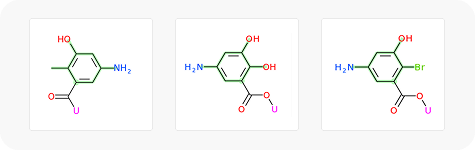
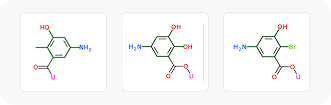
Grow fragments within Enamine REAL and Freedom Space in desired directions. Create focused libraries per hit (up to 100M molecules each).
-
Molecular Docking
Molecular docking of focused libraries and hit selection using molecular mechanics calculations (SQM, mmPBSA, mmGBSA).
-
Synthesis
Synthesis of 100-500 most promising molecules selected in the previous step.
-
Hit Validation
In vitro binding assay(s), cell-based functional assay(s) in DRC (dose-response curve).
REAL Fragment Library
To efficiently grow fragments inside Enamine REAL we designed REAL Fragment Library, a pre-plated 4,960 fragment collection that efficiently covers the chemical space of all synthons inside the REAL Space.
For the creation of this library, all synthons were grouped by their scaffolds, and each group was superimposed in 2D-coordinate space. This allowed us to select the scaffolds that have exit vectors in more than 2 quadrants. It means that scaffolds can be grown in a wider variety of directions. Thus we mapped scaffolds to synthons and selected those that gave us the most chemically diverse compounds.
We offer a final set of 4,960 compounds that are pre-plated and can be ordered from Chemspace. Nevertheless, for a given project customization may be discussed and applied.
REAL Crystallographic Library
Screening fragments using crystallography results in acquiring valuable data on binding modes right after the first screen. However, the main drawback is that it has a much lower throughput. This is why we created a library of 519 compounds based on the REAL Fragment Library for you to be able to benefit from this approach. The compounds were selected using fingerprint-based similarity to ensure their diversity – this way we can provide maximum chemical space coverage with less compounds.
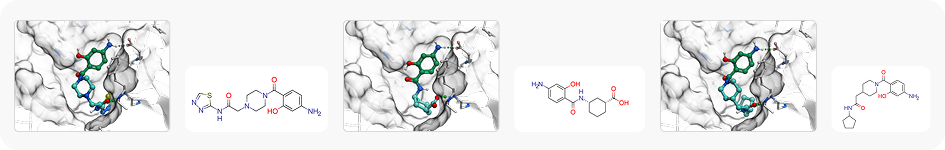
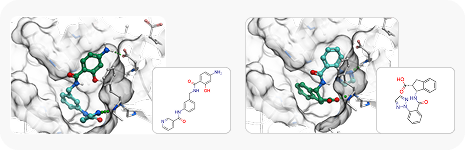
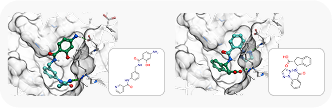
Case Study
To showcase the ability of the proposed approach to efficiently link and grow fragments we applied it to the fragments discovered in the article on the design of binders for Nsp3 macrodomain of SARS-CoV-21. As a result, we have successfully found full molecules with good docking scores for each of the fragments (the docking was performed using ICM-Pro software developed by MolSolf).