Classical Molecular Docking
Classical molecular docking is a computational drug discovery approach that utilizes scoring functions as parameters for prediction of protein-ligand interaction. We perform molecular docking using ICM-Pro software developed by Molsoft. Once the calculations have been accomplished, generated parameters like score function, ML-powered RTCNN (Radial and Topological Convolutional Neural Network Score) score hydrogen bonding are taken into account to select the best binders. A successful attempt to accommodate ligand molecule from big screening collection into a binding pocket of target protein allows to arrive at relatively small selection (100-500 compounds) of good binders that serve an excellent starting point for drug discovery process.
Our Workflow
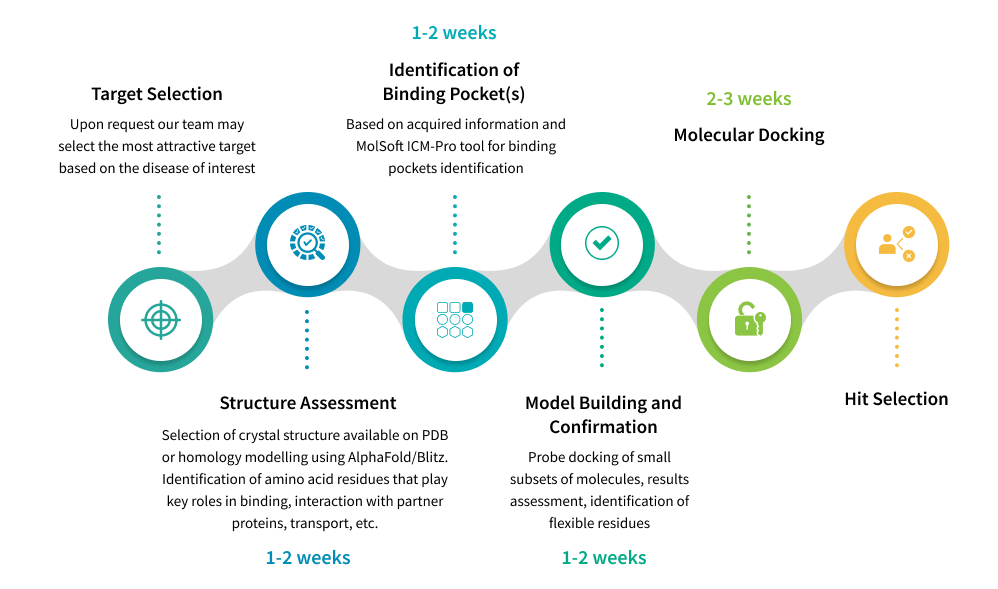
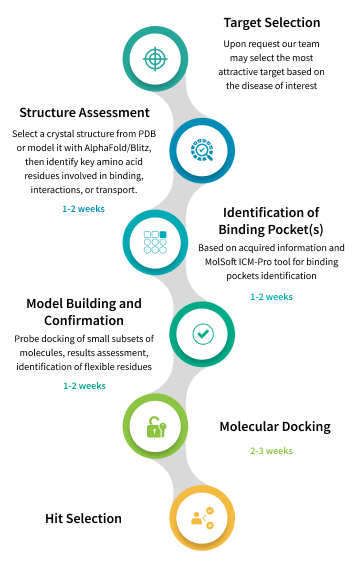
To deliver the most promising compounds to our clients, we stick to the established workflow that reflects the agenda wholly to help our team reduce delays and avert costly rework. At the beginning of drug discovery project we usually receive information about target protein from client, but we can also assist in target selection based on the disease of your interest. After target selection, as the first step our team looks for crystal structure with best resolution available on Protein Data Bank (PDB). If structure of target protein has not been resolved yet, homology modelling using AlphaFold or Blitz may be applied. During this stage we also identify the amino acid residues that play crucial roles in binding, interaction with partner proteins, etc. Next, we utilize tools embedded in our software ICM-Pro developed by MolSoft to identify potential binding pockets. Once a binding pocket has been pinpointed, we build and confirm a model. After completing all the preparatory steps, we perform the classical molecular docking computations with ICM-Pro using in-stock collections of screening compounds provided by Chemspace’s suppliers and Enamine (over 11M compounds total). The obtained data is transferred to hit selection, the most sophisticated stage in molecular docking workflow.
Application of Molecular Docking
Molecular docking is a key tool in structural molecular biology and computer-aided drug design. The concept of this approach is to predict the interaction between a small molecule (ligand) and a protein target, thereby providing the raw material for rational drug design. The applications of molecular docking include:
- Deep understanding of the structural information of a protein of interest with a binding partner.
-
Hit identification
Molecular docking in combination with functional scoring can be used to screen large databases to identify potential drug candidates in silico. - Lead optimization
Molecular docking can be used to predict an optimized orientation of the ligand to the target of interest. Such an approach helps to develop a selective and potent lead compound more efficiently.